Lonestar6 User Guide
Last update: June 9, 2026
Notices
- Queue Restriction: Do not request or exclude specific compute nodes when submitting jobs without prior approval from TACC staff. You must allow Slurm to allocate nodes to your jobs. (06/12/2026)
-
Using Artificial Intelligence (AI) clients on TACC resources: We strongly recommend you run all AI assisted tasks on a compute node. Consult the Good Conduct Guide guide for instructions on the use of AI tools and agents. (05/07/2026)
Important
- You are responsible for any processes launched on TACC resources via any AI process.
- Any and all SUs consumed by an AI process launched by you will be charged to your allocation.
Introduction
Lonestar6 provides a balanced set of resources to support simulation, data analysis, visualization, and machine learning. It is the next system in TACC's Lonestar series of high performance computing systems that are deployed specifically to support Texas researchers. Lonestar6 is funded through collaboration with TACC, the University of Texas System, Texas A&M University, Texas Tech University, and the University of North Texas, as well as a number of research centers and faculty at UT-Austin, including the Oden Institute for Computational Engineering & Sciences and the Center for Space Research.
The system employs Dell Servers with AMD's highly performant Epyc Milan processor, Mellanox's HDR Infiniband technology, and 8 PB of BeeGFS based storage on Dell storage hardware. Additionally, Lonestar6 supports GPU nodes utilizing NVIDIA's A100 and H100 GPUs to support machine learning workflows and other GPU-enabled applications. Lonestar6 will continue to support the TACC HPC environment, providing numerical libraries, parallel applications, programming tools, and performance monitoring capabilities to the user community.
System Architecture
All Lonestar6 nodes run Rocky 8.4 and are managed with batch services through native Slurm 20.11.8. Global storage areas are supported by an NFS file system ($HOME), a BeeGFS parallel file system ($SCRATCH), and a Lustre parallel file system ($WORK). Inter-node communication is supported by a Mellanox HDF Infiniband network. Also, the TACC Ranch tape archival system is available from Lonestar6.
The system is composed of 560 compute nodes and 88 GPU nodes: 84 A100 GPU nodes and 4 H100 nodes with 2 NVIDIA H100 GPUs each. The compute nodes are housed in 4 dielectric liquid coolant cabinets and ten air-cooled racks. The air cooled racks also contain the 88 GPU nodes. Each node has two AMD EPYC 7763 64-core processors (Milan) and 256 GB of DDR4 memory. Twenty-four of the compute nodes are reserved for development and are accessible interactively for up to two hours. Each of the system's 84 A100 GPU nodes also contains two AMD EPYC 7763 64-core processes and three NVIDIA A100 GPUs each with 40 GB of high bandwidth memory (HBM2).
Compute Nodes
Lonestar6 hosts 560 compute nodes with 5 TFlops of peak performance per node and 256 GB of DRAM.
Table 1. Compute Node Specifications
| Specification | Value |
|---|---|
| CPU: | 2x AMD EPYC 7763 64-Core Processor ("Milan") |
| Total cores per node: | 128 cores on two sockets (64 cores / socket ) |
| Hardware threads per core: | 1 per core |
| Hardware threads per node: | 128 x 1 = 128 |
| Clock rate: | 2.45 GHz (Boost up to 3.5 GHz) |
| RAM: | 256 GB (3200 MT/s) DDR4 |
| Cache: | 32KB L1 data cache per core 512KB L2 per core 32 MB L3 per core complex (1 core complex contains 8 cores) 256 MB L3 total (8 core complexes ) Each socket can cache up to 288 MB (sum of L2 and L3 capacity) |
| Local storage: | 288GB /tmp partition on a 288GB SSD. |
Login Nodes
Lonestar6's three login nodes, login1, login2, and login3, contain the same hardware and are configured similarly to the compute nodes. However, since these nodes are shared, limits are enforced on memory usage and number of processes. Please use the login nodes only for file management, compilation, and data movement. Any and all computing should be done within a batch job or an interactive session on the compute nodes. See TACC's Good Conduct Policy for more information.
vm-small Queue Nodes
Lonestar6 hosts 28 vm-small compute nodes running on 4 physical hosts.
Table 1.5. vm-small Compute Node Specifications
| Specification | Value |
|---|---|
| CPU: | 1/4th of an AMD EPYC 7763 64-Core Processor ("Milan") |
| Total cores per VM: | 16 cores |
| Hardware threads per core: | 1 per core |
| Hardware threads per VM: | 16 x 1 = 16 |
| Clock rate: | 2.45 GHz (Boost up to 3.5 GHz) |
| RAM: | 32 GB (3200 shared MT/s) DDR4 |
| Cache: | Shared caches with all other VMs. 32KB L1 data cache per core 512KB L2 per core 32 MB L3 per core complex (1 core complex contains 8 cores) 64 MB L3 total (2 core complexes) |
| Local storage: | 288G /tmp partition |
GPU Nodes
Lonestar6 hosts 84 A100 GPU nodes that are configured identically to the compute nodes with the addition of 3 NVIDIA A100 GPUs. Each A100 GPU has a peak performance of 9.7 TFlops in double precision and 312 TFlops in FP16 precision using the Tensor Cores. Additionally, there are 4 H100 GPU nodes that support 2 NVIDIA H100 GPUs. Each H100 GPU has a peak performance of 26 TFlops in double precision and 1513 TFlops in FP16 precision using the Tensor cores.
Table 2. A100 GPU Node Specifications
| Specification | Value |
|---|---|
| GPU: | 3x NVIDIA A100 PCIE 40GB gpu0: socket 0 gpu1: socket1 gpu2: socket1 |
| GPU Memory: | 40 GB HBM2 |
| CPU: | 2x AMD EPYC 7763 64-Core Processor ("Milan") |
| Total cores per node: | 128 cores on two sockets (64 cores / socket ) |
| Hardware threads per core: | 1 per core |
| Hardware threads per node: | 128 x 1 = 128 |
| Clock rate: | 2.45 GHz |
| RAM: | 256 GB |
| Cache: | 32KB L1 data cache per core 512KB L2 per core 32 MB L3 per core complex (1 core complex contains 8 cores) 256 MB L3 total (8 core complexes ) Each socket can cache up to 288 MB (sum of L2 and L3 capacity) |
| Local storage: | 288GB /tmp partition |
Table 2.5 H100 GPU Node Specifications
| Specification | Value |
|---|---|
| GPU: | 2x NVIDIA H100 PCIE 80GB gpu0: socket 0 gpu1: socket 1 |
| GPU Memory: | 80 GB HBM2e |
| CPU: | 2x AMD EPYC 9454 48-Core Processor ("Genoa") |
| Total cores per node: | 96 cores on two sockets (48 cores / socket ) |
| Hardware threads per core: | 1 per core |
| Hardware threads per node: | 96 x 1 = 96 |
| Clock rate: | 2.75 GHz |
| RAM: | 384 GB |
| Cache: | 64KB L1 data cache per core 1MB L2 per core 32 MB L3 per core complex (1 core complex contains 8 cores) 256 MB L3 total (8 core complexes ) Each socket can cache up to 304 MB (sum of L2 and L3 capacity) |
| Local storage: | 288GB /tmp partition |
Network
The interconnect is based on Mellanox HDR technology with full HDR (200 Gb/s) connectivity between the switches and the compute nodes. A fat tree topology employing sixteen core switches connects the compute nodes and the $SCRATCH file systems. There is an oversubscription of 24/16.
Allocations
Lonestar6 is available to researchers from all University of Texas System institutions and to our partners, Texas A&M University, Texas Tech University and University of North Texas.
UT System Researchers may submit allocation requests for compute time on Lonestar6 via TACC's new Texas Resource Allocation System (TxRAS). Consult the Allocations page for details.
Researchers at our partner institutions may submit allocation requests through the links below.
Important
You must be added to a Lonestar6 allocation in order to have access/login to Lonestar6.
Monitor Allocation Usage
Once you've been awarded or added to an allocation, you may monitor usage in one of two ways:
-
The
taccinfocommand will display a summary of your TACC project balances and disk quotas at any time. Invoke the command using the absolute path as shown below. This output is generally more current than balances displayed on the portals.login1$ /usr/local/etc/taccinfo --------------------- Project balances for user bjones ------------------------ | Name Avail SUs Expires | Name Avail SUs Expires | | A-ccsc 342808 2030-09-30 | A-cciq 4828 2026-09-30 | ------------------------ Disk quotas for user blones -------------------------- | Disk Usage (GB) Limit %Used File Usage Limit %Used | | /scratch 6.6 0.0 0.00 22742 0 0.00 | | /home1 2.5 10.0 25.07 18488 0 0.00 | | /work 1.1 1024.0 0.11 23890 3000000 0.80 | ------------------------------------------------------------------------------- -
Principal Investigators can monitor allocation usage via the TACC User Portal under "Allocations->Projects and Allocations". Be aware that the figures shown on the portal may lag behind the most recent usage.
Access the System
Secure Shell (SSH)
The ssh command (SSH protocol) is the standard way to connect to Lonestar6 (ls6.tacc.utexas.edu). SSH also includes support for the file transfer utilities scp and sftp. Wikipedia is a good source of information on SSH. SSH is available within Linux and from the terminal app in the Mac OS. If you are using Windows, you will need an SSH client that supports the SSH-2 protocol: e.g. Bitvise, OpenSSH, PuTTY, or SecureCRT. Initiate a session using the ssh command or the equivalent; from the Linux command line the launch command looks like this:
localhost$ ssh username@ls6.tacc.utexas.eduThe above command will rotate connections across all available login nodes, login1-login3, and route your connection to one of them. To connect to a specific login node, use its full domain name:
localhost$ ssh username@login2.ls6.tacc.utexas.eduTo connect with X11 support on Lonestar6 (usually required for applications with graphical user interfaces), use the -X or -Y switch:
localhost$ ssh -X username@ls6.tacc.utexas.eduTo report a connection problem, execute the ssh command with the -vvv option and include the verbose output when submitting a help ticket.
Do not run the ssh-keygen command on Lonestar6. This command will create and configure a key pair that will interfere with the execution of job scripts in the batch system. If you do this by mistake, you can recover by renaming or deleting the .ssh directory located in your home directory; the system will automatically generate a new one for you when you next log into Lonestar6.
- execute
mv .ssh dot.ssh.old - log out
- log into Lonestar6 again
After logging in again the system will generate a properly configured key pair.
Regardless of your research workflow, you'll need to master Linux basics and a Linux-based text editor (e.g. emacs, nano, gedit, or vi/vim) to use the system properly. However, this user guide does not address these topics. There are numerous resources in a variety of formats that are available to help you learn Linux. If you encounter a term or concept in this user guide that is new to you, a quick internet search should help you resolve the matter quickly.
Linux Shell
The default login shell for your user account is Bash. To determine your current login shell, execute:
$ echo $SHELLIf you'd like to change your login shell to csh, sh, tcsh, or zsh, submit a helpdesk ticket. The chsh ("change shell") command will not work on TACC systems.
When you start a shell on Lonestar6, system-level startup files initialize your account-level environment and aliases before the system sources your own user-level startup scripts. You can use these startup scripts to customize your shell by defining your own environment variables, aliases, and functions. These scripts (e.g. .profile and .bashrc) are generally hidden files: so-called dotfiles that begin with a period, visible when you execute: ls -a.
Before editing your startup files, however, it's worth taking the time to understand the basics of how your shell manages startup. Bash startup behavior is very different from the simpler csh behavior, for example. The Bash startup sequence varies depending on how you start the shell (e.g. using ssh to open a login shell, executing the bash command to begin an interactive shell, or launching a script to start a non-interactive shell). Moreover, Bash does not automatically source your .bashrc when you start a login shell by using ssh to connect to a node. Unless you have specialized needs, however, this is undoubtedly more flexibility than you want: you will probably want your environment to be the same regardless of how you start the shell. The easiest way to achieve this is to execute source ~/.bashrc from your .profile, then put all your customizations in .bashrc. The system-generated default startup scripts demonstrate this approach. We recommend that you use these default files as templates.
For more information see the Bash Users' Startup Files: Quick Start Guide and other online resources that explain shell startup. To recover the originals that appear in a newly created account, execute /usr/local/startup_scripts/install_default_scripts.
Environment Variables
Your environment includes the environment variables and functions defined in your current shell: those initialized by the system, those you define or modify in your account-level startup scripts, and those defined or modified by the modules that you load to configure your software environment. Be sure to distinguish between an environment variable's name (e.g. HISTSIZE) and its value ($HISTSIZE). Understand as well that a sub-shell (e.g. a script) inherits environment variables from its parent, but does not inherit ordinary shell variables or aliases. Use export (in Bash) or setenv (in csh) to define an environment variable.
Execute the env command to see the environment variables that define the way your shell and child shells behave.
Pipe the results of env into grep to focus on specific environment variables. For example, to see all environment variables that contain the string GIT (in all caps), execute:
$ env | grep GITThe environment variables PATH and LD_LIBRARY_PATH are especially important. PATH is a colon-separated list of directory paths that determines where the system looks for your executables. LD_LIBRARY_PATH is a similar list that determines where the system looks for shared libraries.
Account-Level Diagnostics
TACC's sanitytool module loads an account-level diagnostic package that detects common account-level issues and often walks you through the fixes. You should certainly run the package's sanitycheck utility when you encounter unexpected behavior. You may also want to run sanitycheck periodically as preventive maintenance. To run sanitytool's account-level diagnostics, execute the following commands:
login1$ module load sanitytool
login1$ sanitycheckExecute module help sanitytool for more information.
Using Modules to Manage your Environment
Lmod, a module system developed and maintained at TACC, makes it easy to manage your environment so you have access to the software packages and versions that you need to conduct your research. This is especially important on a system like Lonestar6 that serves thousands of users with an enormous range of needs. Loading a module amounts to choosing a specific package from among available alternatives:
$ module load intel # load the default Intel compiler v19.1.14
$ module load intel/19.1.1 # load a specific version of the Intel compilerA module does its job by defining or modifying environment variables (and sometimes aliases and functions). For example, a module may prepend appropriate paths to $PATH and $LD_LIBRARY_PATH so that the system can find the executables and libraries associated with a given software package. The module creates the illusion that the system is installing software for your personal use. Unloading a module reverses these changes and creates the illusion that the system just uninstalled the software:
$ module load netcdf # defines DDT-related env vars; modifies others
$ module unload netcdf # undoes changes made by loadThe module system does more, however. When you load a given module, the module system can automatically replace or deactivate modules to ensure the packages you have loaded are compatible with each other. In the example below, the module system automatically unloads one compiler when you load another, and replaces Intel-compatible versions of IMPI and FFTW3 with versions compatible with gcc:
$ module load intel # load default version of Intel compiler
$ module load fftw3 # load default version of fftw3
$ module load gcc # change compiler
Lmod is automatically replacing "intel/19.0.4" with "gcc/9.1.0".
Inactive Modules:
1) python2
Due to MODULEPATH changes, the following have been reloaded:
1) fftw3/3.3.8 2) impi/19.0.4On Lonestar6, modules generally adhere to a TACC naming convention when defining environment variables that are helpful for building and running software. For example, the papi module defines TACC_PAPI_BIN (the path to PAPI executables), TACC_PAPI_LIB (the path to PAPI libraries), TACC_PAPI_INC (the path to PAPI include files), and TACC_PAPI_DIR (top-level PAPI directory). After loading a module, here are some easy ways to observe its effects:
$ module show netcdf # see what this module does to your environment
$ env | grep NETCDF # see env vars that contain the string PAPI
$ env | grep -i netcdf # case-insensitive search for 'papi' in environmentTo see the modules you currently have loaded:
$ module listTo see all modules that you can load right now because they are compatible with the currently loaded modules:
$ module availTo see all installed modules, even if they are not currently available because they are incompatible with your currently loaded modules:
$ module spider # list all modules, even those not available to loadTo filter your search:
$ module spider netcdf # all modules with names containing 'slep'
$ module spider netcdf/3.6.3 # additional details on a specific moduleAmong other things, the latter command will tell you which modules you need to load before the module is available to load. You might also search for modules that are tagged with a keyword related to your needs (though your success here depends on the diligence of the module writers). For example:
$ module keyword performanceYou can save a collection of modules as a personal default collection that will load every time you log into Lonestar6. To do so, load the modules you want in your collection, then execute:
$ module save # save the currently loaded collection of modules Two commands make it easy to return to a known, reproducible state:
$ module reset # load the system default collection of modules
$ module restore # load your personal default collection of modulesOn TACC systems, the command module reset is equivalent to module purge; module load TACC. It's a safer, easier way to get to a known baseline state than issuing the two commands separately.
Help text is available for both individual modules and the module system itself:
$ module help swr # show help text for software package swr
$ module help # show help text for the module system itselfSee Lmod's online documentation for more extensive documentation. The online documentation addresses the basics in more detail, but also covers several topics beyond the scope of the help text (e.g. writing and using your own module files).
It's safe to execute module commands in job scripts. In fact, this is a good way to write self-documenting, portable job scripts that produce reproducible results. If you use module save to define a personal default module collection, it's rarely necessary to execute module commands in shell startup scripts, and it can be tricky to do so safely. If you do wish to put module commands in your startup scripts, see Lonestar6's default startup scripts for a safe way to do so.
Crontabs
TACC allows cronjobs but be aware that crontab files are unique to the login node where they were created and are not shared across the login nodes. Crontab files are not allowed on the compute nodes.
Note
All TACC HPC systems host multiple login nodes. When you login, your connection is routed to the next available login node via round-robin DNS. This practice balances the user load across the system.
When creating a crontab file, use the hostname command to determine your exact location, and make note of it:
$ hostname
login2.lonestar6.tacc.utexas.eduSimilarly you can always connect to that login node by specifying its full domain name:
localhost$ ssh login2.lonestar6.tacc.utexas.eduImportant
As with any computation, ensure that cronjobs are run only on the compute nodes
TACC Tips
TACC staff has amassed a database of helpful tips for our users. Access these tips via the tacc_tips module and showTip command as demonstrated below:
$ module load tacc_tips
$ showTip
Tip 40 (See "module help tacc_tips" for features or how to disable)
Here are four different ways to repeat the last executed command (press enter after each):
* Use the up arrow
* Type !!
* Type !-1
* Press Ctrl+PManaging Files
Tip
Each "Active" TACC account user is allocated 1TB of space across the shared /work (Stockyard) file system, as well as 2TB on TACC's archival system, Ranch.
Table 3. File Systems
| File System | Quota | Key Features |
|---|---|---|
$HOME |
10 GB | 200,000 files Not intended for parallel or high-intensity file operations. NFS file system Backed up regularly. Overall capacity 7 TB Not purged. |
$WORK |
1 TB 3,000,000 files Across all TACC systems |
Not intended for high-intensity file operations or jobs involving very large files. Lustre file system On the Global Shared File System that is mounted on most TACC systems. See Stockyard system description for more information. Defaults: 1 stripe, 1MB stripe size Not backed up. Not purged. |
$SCRATCH |
none | Overall capacity 8 PB Defaults: 4 targets, 512 KB chunk size Not backed up Files are subject to purge if access time* is more than 10 days old.* |
/tmp on nodes |
288 GB | Data purged at the end of each job. Access is local to the node. Data in /tmp is not shared across nodes. |
Important
TACC will not grant requests for increasing /home and /work quotas. If you require more storage space, TACC's Corral is available at no charge to UT researchers, and at low-cost annual fee to non-UT researchers. See the Corral User Guide for more details.
Scratch File System Purge Policy
Warning
The $SCRATCH file system, as its name indicates, is a temporary storage space. Files that have not been accessed* in ten days are subject to purge.
Deliberately modifying file access time, using any method, tool, or program, for the purpose of circumventing purge policies is prohibited.
*The operating system updates a file's access time when that file is modified on a login or compute node. Reading or executing a file/script on a login node does not update the access time, but reading or executing on a compute node does update the access time. This approach helps us distinguish between routine management tasks (e.g. tar, scp) and production use. Use the command ls -ul to view access times.
Navigating the Shared File Systems
Lonestar6 mounts three Lustre file systems that are shared across all nodes: the home, work, and scratch file systems. Lonestar6's startup mechanisms define corresponding account-level environment variables $HOME, $SCRATCH and $WORK that store the paths to directories that you own on each of these file systems. Consult the Lonestar6 File Systems table above for the basic characteristics of these file systems, and the Good Conduct document for guidance on file system etiquette.
Lonestar6's /home and /scratch file systems are mounted only on Lonestar6, but the /work file system mounted on Lonestar6 is the Global Shared File System hosted on Stockyard. This is the same work file system that is currently available on Frontera, Stampede3 and most other TACC resources.
The $STOCKYARD environment variable points to the highest-level directory that you own on the Global Shared File System. The definition of the $STOCKYARD environment variable is of course account-specific, but you will see the same value on all TACC systems that provide access to the Global Shared File System (see Table 3). This directory is an excellent place to store files you want to access regularly from multiple TACC resources.
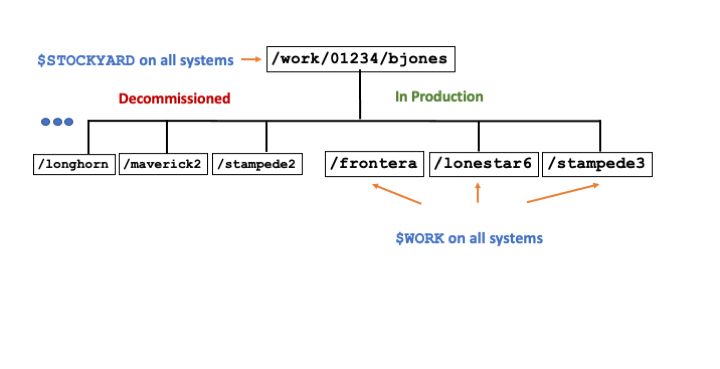
/work file system (Global Shared File System hosted on Stockyard). Example for fictitious user bjones. All directories usable from all systems. Sub-directories (e.g. stampede3, frontera) exist only when you have allocations on the associated system.Your account-specific $WORK environment variable varies from system to system and is a subdirectory of $STOCKYARD (Figure 1). The subdirectory name corresponds to the associated TACC resource. The $WORK environment variable on Lonestar6 points to the $STOCKYARD/ls6 subdirectory, a convenient location for files you use and jobs you run on Lonestar6. Remember, however, that all subdirectories contained in your $STOCKYARD directory are available to you from any system that mounts the file system. If you have accounts on both Lonestar6 and Stampede3, for example, the $STOCKYARD/ls6 directory is available from your Stampede3 account, and $STOCKYARD/stampede3 directory is available from your Lonestar6 account. Your quota and reported usage on the Global Shared File System reflects all files that you own on Stockyard, regardless of their actual location on the file system.
Note that resource-specific subdirectories of $STOCKYARD are simply convenient ways to manage your resource-specific files. You have access to any such subdirectory from any TACC resources. If you are logged into Lonestar6, for example, executing the alias cdw (equivalent to cd $WORK) will take you to the resource-specific subdirectory $STOCKYARD/ls6. But you can access this directory from other TACC systems as well by executing cd $STOCKYARD/ls6. These commands allow you to share files across TACC systems. In fact, several convenient account-level aliases make it even easier to navigate across the directories you own in the shared file systems:
Table 4. Built-in Account Level Aliases
| Alias | Command |
|---|---|
cd or cdh |
cd $HOME |
cds |
cd $SCRATCH |
cdy or cdg |
cd $STOCKYARD |
cdw |
cd $WORK |
Striping Large Files
Lonestar6's BeeGFS and Lustre file systems look and act like a single logical hard disk, but are actually sophisticated integrated systems involving many physical drives. Lustre and BeeGFS can stripe (distribute 'chunk's) large files over several physical disks, making it possible to deliver the high performance needed to service input/output (I/O) requests from hundreds of users across thousands of nodes. Object Storage Targets (OSTs) manage the file system's spinning disks: a file with 16 stripes, for example, is distributed across 16 OSTs. One designated Meta-Data Server (MDS) tracks the OSTs assigned to a file, as well as the file's descriptive data.
The Lonestar6 $SCRATCH filesystem is a BeeGFS filesystem instead of a Lustre filesystem. However, the BeeGFS filesystem is similar to Lustre in that it distributes files across I/O servers and allows the user control over the stripe count and stripe size for directories.
The $WORK file system has 24 I/O targets available, while the $SCRATCH file system has 96. A good rule of thumb is to allow at least one stripe for each 100GB in the file not to exceed 75% of the available stripes. So, the max stripe count for $SCRATCH would be 72, while that for $WORK would be 18.
Important
Before transferring to, or creating large files on Lonestar6, be sure to set an appropriate default chunk/stripe count on the receiving directory.
As an example, the following command sets the default stripe count on the current directory for a file 200 GB in size, then ensures that the operation was successful:
-
If the destination directory is on the
$SCRATCHfile system:$ beegfs-ctl --setpattern --numtargets=20 $PWD $ beegfs-ctl --getentryinfo $PWD -
If the destination directory is on the
$WORKfile system:$ lfs setstripe -c 18 $PWD #Use stripe count of 18 instead of 20 out of 24 total targets $ lfs getstripe $PWD
Important
It is not possible to change the stripe/chunk count on a file that already exists. The mv command will have no effect on a file's striping unless the source and destination directories are on different file systems, e.g. mving a file from $SCRATCH to $WORK, or vice-versa. Instead, use the cp command to copy the file to a directory with the intended stripe parameters.
Run the following for more information on these commands:
$ beegfs-ctl --setpattern --help
$ lfs help setstripe
Transferring your Files
Transferring with scp
You can transfer files between Lonestar6 and Linux-based systems using either scp or rsync. Both scp and rsync are available in the Mac Terminal app. Windows SSH clients typically include scp-based file transfer capabilities.
The Linux scp (secure copy) utility is a component of the OpenSSH suite. Assuming your Lonestar6 username is bjones, a simple scp transfer that pushes a file named myfile from your local Linux system to Lonestar6 $HOME would look like this:
localhost$ scp ./myfile bjones@ls6.tacc.utexas.edu: # note colon after net addressYou can use wildcards, but you need to be careful about when and where you want wildcard expansion to occur. For example, to push all files ending in .txt from the current directory on your local machine to /work/01234/bjones/scripts on Lonestar6:
localhost$ scp *.txt bjones@ls6.tacc.utexas.edu:/work/01234/bjones/ls6To delay wildcard expansion until reaching Lonestar6, use a backslash (\) as an escape character before the wildcard. For example, to pull all files ending in .txt from /work/01234/bjones/scripts on Lonestar6 to the current directory on your local system:
localhost$ scp bjones@ls6.tacc.utexas.edu:/work/01234/bjones/ls6/\*.txt .Note
Using scp with wildcard expansion on the remote host is unreliable. Specify absolute paths wherever possible.
Avoid using scp for recursive transfers of directories that contain nested directories of many small files:
localhost$ scp -r ./mydata bjones@ls6.tacc.utexas.edu:\$SCRATCH # DON'T DO THISInstead, use tar to create an archive of the directory, then transfer the directory as a single file:
localhost$ tar cvf ./mydata.tar mydata # create archive
localhost$ scp ./mydata.tar bjones@ls6.tacc.utexas.edu:\$WORK # transfer archiveTransferring with rsync
The rsync (remote synchronization) utility is a great way to synchronize files that you maintain on more than one system: when you transfer files using rsync, the utility copies only the changed portions of individual files. As a result, rsync is especially efficient when you only need to update a small fraction of a large dataset. The basic syntax is similar to scp:
localhost$ rsync mybigfile bjones@ls6.tacc.utexas.edu:\$SCRATCH/data
localhost$ rsync -avtr mybigdir bjones@ls6.tacc.utexas.edu:\$SCRATCH/dataThe options on the second transfer are typical and appropriate when synching a directory: this is a recursive update (-r) with verbose (-v) feedback; the synchronization preserves time stamps (-t) as well as symbolic links and other meta-data (-a). Because rsync only transfers changes, recursive updates with rsync may be less demanding than an equivalent recursive transfer with scp.
Sharing Files with Collaborators
If you wish to share files and data with collaborators in your project, see Sharing Project Files on TACC Systems for step-by-step instructions. Project managers or delegates can use Unix group permissions and commands to create read-only or read-write shared workspaces that function as data repositories and provide a common work area to all project members.
Building Software
Important
TACC maintains a database of currently installed software packages and libraries across all HPC resources. Navigate to TACC's Software List to see where, or if, a particular package is already installed on a particular resource.
If TACC does not have your desired software package already installed, you are welcome to download, build, and install the package in your own account. See Building Third-Party Software in the Software at TACC guide.
The phrase "building software" is a common way to describe the process of producing a machine-readable executable file from source files written in C, Fortran, or some other programming language. In its simplest form, building software involves a simple, one-line call or short shell script that invokes a compiler. More typically, the process leverages the power of makefiles, so you can change a line or two in the source code, then rebuild in a systematic way only the components affected by the change. Increasingly, however, the build process is a sophisticated multi-step automated workflow managed by a special framework like autotools or cmake, intended to achieve a repeatable, maintainable, portable mechanism for installing software across a wide range of target platforms.
Basics of Building Software
This section of the user guide does nothing more than introduce the big ideas with simple one-line examples. You will undoubtedly want to explore these concepts more deeply using online resources. You will quickly outgrow the examples here. We recommend that you master the basics of makefiles as quickly as possible: even the simplest computational research project will benefit enormously from the power and flexibility of a makefile-based build process.
Intel Compilers
Intel is the recommended and default compiler suite on Lonestar6. Each Intel module also gives you direct access to mkl without loading an mkl module; see Intel MKL for more information. Here are simple examples that use the Intel compiler to build an executable from source code:
Compiling a code that uses OpenMP would look like this:
$ icc -qopenmp mycode.c -o myexe # OpenMPSee the published Intel documentation, available both online and in ${TACC_INTEL_DIR}/documentation, for information on optimization flags and other Intel compiler options.
GNU Compilers
The GNU foundation maintains a number of high quality compilers, including a compiler for C (gcc), C++ (g++), and Fortran (gfortran). The gcc compiler is the foundation underneath all three, and the term gcc often means the suite of these three GNU compilers.
Load a gcc module to access a recent version of the GNU compiler suite. Avoid using the GNU compilers that are available without a gcc module — those will be older versions based on the "system gcc" that comes as part of the Linux distribution.
Here are simple examples that use the GNU compilers to produce an executable from source code:
$ gcc mycode.c # C source file; executable a.out
$ gcc mycode.c -o myexe # C source file; executable myexe
$ g++ mycode.cpp -o myexe # C++ source file
$ gfortran mycode.f90 -o myexe # Fortran90 source file
$ gcc -fopenmp mycode.c -o myexe # OpenMP; GNU flag is different than IntelNote that some compiler options are the same for both Intel and GNU (e.g. -o), while others are different (e.g. -qopenmp vs -fopenmp). Many options are available in one compiler suite but not the other. See the online GNU documentation for information on optimization flags and other GNU compiler options.
Compiling and Linking as Separate Steps
Building an executable requires two separate steps: (1) compiling (generating a binary object file associated with each source file); and (2) linking (combining those object files into a single executable file that also specifies the libraries that executable needs). The examples in the previous section accomplish these two steps in a single call to the compiler. When building more sophisticated applications or libraries, however, it is often necessary or helpful to accomplish these two steps separately.
Use the -c ("compile") flag to produce object files from source files:
$ icc -c main.c calc.c results.cBarring errors, this command will produce object files main.o, calc.o, and results.o. Syntax for other compilers Intel and GNU compilers is similar.
You can now link the object files to produce an executable file:
$ icc main.o calc.o results.o -o myexeThe compiler calls a linker utility (usually /bin/ld) to accomplish this task. Again, syntax for other compilers is similar.
Include and Library Paths
Software often depends on pre-compiled binaries called libraries. When this is true, compiling usually requires using the -I option to specify paths to so-called header or include files that define interfaces to the procedures and data in those libraries. Similarly, linking often requires using the -L option to specify paths to the libraries themselves. Typical compile and link lines might look like this:
$ icc -c main.c -I${WORK}/mylib/inc -I${TACC_HDF5_INC} # compile
$ icc main.o -o myexe -L${WORK}/mylib/lib -L${TACC_HDF5_LIB} -lmylib -lhdf5 # linkOn Lonestar6, both the hdf5 and phdf5 modules define the environment variables $TACC_HDF5_INC and $TACC_HDF5_LIB. Other module files define similar environment variables; see Using Modules for more information.
The details of the linking process vary, and order sometimes matters. Much depends on the type of library: static (.a suffix; library's binary code becomes part of executable image at link time) versus dynamically-linked shared (.so suffix; library's binary code is not part of executable; it's located and loaded into memory at run time). The link line can use rpath to store in the executable an explicit path to a shared library. In general, however, the LD_LIBRARY_PATH environment variable specifies the search path for dynamic libraries. For software installed at the system-level, TACC's modules generally modify LD_LIBRARY_PATH automatically. To see whether and how an executable named myexe resolves dependencies on dynamically linked libraries, execute ldd myexe.
A separate section below addresses the Intel Math Kernel Library (MKL).
Compiling and Linking MPI Programs
Intel MPI (module impi) and MVAPICH2 (module mvapich2) are the two MPI libraries available on Lonestar6. After loading an impi or mvapich2 module, compile and/or link using an mpi wrapper (mpicc, mpicxx, mpif90) in place of the compiler:
$ mpicc mycode.c -o myexe # C source, full build
$ mpicc -c mycode.c # C source, compile without linking
$ mpicxx mycode.cpp -o myexe # C++ source, full build
$ mpif90 mycode.f90 -o myexe # Fortran source, full buildThese wrappers call the compiler with the options, include paths, and libraries necessary to produce an MPI executable using the MPI module you're using. To see the effect of a given wrapper, call it with the -show option:
$ mpicc -show # Show compile line generated by call to mpicc; similarly for other wrappersIntel Math Kernel Library (MKL)
The Intel Math Kernel Library (MKL) is a collection of highly optimized functions implementing some of the most important mathematical kernels used in computational science, including standardized interfaces to:
- BLAS (Basic Linear Algebra Subroutines), a collection of low-level matrix and vector operations like matrix-matrix multiplication
- LAPACK (Linear Algebra PACKage), which includes higher-level linear algebra algorithms like Gaussian Elimination
- FFT (Fast Fourier Transform), including interfaces based on FFTW (Fastest Fourier Transform in the West)
- ScaLAPACK (Scalable LAPACK), BLACS (Basic Linear Algebra Communication Subprograms), Cluster FFT, and other functionality that provide block-based distributed memory (multi-node) versions of selected LAPACK, BLAS, and FFT algorithms;
- Vector Mathematics (VM) functions that implement highly optimized and vectorized versions of special functions like sine and square root.
MKL with Intel C, C++, and Fortran Compilers
There is no MKL module for the Intel compilers because you don't need one: the Intel compilers have built-in support for MKL. Unless you have specialized needs, there is no need to specify include paths and libraries explicitly. Instead, using MKL with the Intel modules requires nothing more than compiling and linking with the -mkl option.; e.g.
$ icc -mkl mycode.c
$ ifort -mkl mycode.cThe -mkl switch is an abbreviated form of -mkl=parallel, which links your code to the threaded version of MKL. To link to the unthreaded version, use -mkl=sequential. A third option, -mkl=cluster, which also links to the unthreaded libraries, is necessary and appropriate only when using ScaLAPACK or other distributed memory packages. For additional information, including advanced linking options, see the MKL documentation and Intel MKL Link Line Advisor.
MKL with GNU C, C++, and Fortran Compilers
When using a GNU compiler, load the MKL module before compiling or running your code, then specify explicitly the MKL libraries, library paths, and include paths your application needs. Consult the Intel MKL Link Line Advisor for details. A typical compile/link process on a TACC system will look like this:
$ module load gcc
$ module load mkl # available/needed only for GNU compilers
$ gcc -fopenmp -I$MKLROOT/include \
-Wl,-L${MKLROOT}/lib/intel64 \
-lmkl_intel_lp64 -lmkl_core \
-lmkl_gnu_thread -lpthread \
-lm -ldl mycode.cFor your convenience the mkl module file also provides alternative TACC-defined variables like $TACC_MKL_INCLUDE (equivalent to $MKLROOT/include). Execute module help mkl for more information.
Using MKL as BLAS/LAPACK with Third-Party Software
When your third-party software requires BLAS or LAPACK, you can use MKL to supply this functionality. Replace generic instructions that include link options like -lblas or -llapack with the simpler MKL approach described above. There is no need to download and install alternatives like OpenBLAS.
Using MKL as BLAS/LAPACK with TACC's MATLAB, Python, and R Modules
TACC's MATLAB, Python, and R modules all use threaded (parallel) MKL as their underlying BLAS/LAPACK library. These means that even serial codes written in MATLAB, Python, or R may benefit from MKL's thread-based parallelism. This requires no action on your part other than specifying an appropriate max thread count for MKL; see the section below for more information.
Controlling Threading in MKL
Any code that calls MKL functions can potentially benefit from MKL's thread-based parallelism; this is true even if your code is not otherwise a parallel application. If you are linking to the threaded MKL (using -mkl, -mkl=parallel, or the equivalent explicit link line), you need only specify an appropriate value for the max number of threads available to MKL. You can do this with either of the two environment variables MKL_NUM_THREADS or OMP_NUM_THREADS. The environment variable MKL_NUM_THREADS specifies the max number of threads available to each instance of MKL, and has no effect on non-MKL code. If MKL_NUM_THREADS is undefined, MKL uses OMP_NUM_THREADS to determine the max number of threads available to MKL functions. In either case, MKL will attempt to choose an optimal thread count less than or equal to the specified value. Note that OMP_NUM_THREADS defaults to 1 on TACC systems; if you use the default value you will get no thread-based parallelism from MKL.
If you are running a single serial, unthreaded application (or an unthreaded MPI code involving a single MPI task per node) it is usually best to give MKL as much flexibility as possible by setting the max thread count to the total number of hardware threads on the node (128 on AMD Milan). Of course things are more complicated if you are running more than one process on a node: e.g. multiple serial processes, threaded applications, hybrid MPI-threaded applications, or pure MPI codes running more than one MPI rank per node. See http://software.intel.com/en-us/articles/recommended-settings-for-calling-intel-mkl-routines-from-multi-threaded-applications and related Intel resources for examples of how to manage threading when calling MKL from multiple processes.
Using ScaLAPACK, Cluster FFT, and Other MKL Cluster Capabilities
See "Working with the Intel Math Kernel Library Cluster Software" and "Intel MKL Link Line Advisor" for information on linking to the MKL cluster components.
Launching Applications
The primary purpose of your job script is to launch your research application. How you do so depends on several factors, especially (1) the type of application (e.g. MPI, OpenMP, serial), and (2) what you're trying to accomplish (e.g. launch a single instance, complete several steps in a workflow, run several applications simultaneously within the same job). While there are many possibilities, your own job script will probably include a launch line that is a variation of one of the examples described in this section.
One Serial Application
To launch a serial application, simply call the executable. Specify the path to the executable in either the PATH environment variable or in the call to the executable itself:
myprogram # executable in a directory listed in $PATH
$SCRATCH/apps/mydir/myprogram # explicit full path to executable
./myprogram # executable in current directory
./myprogram -m -k 6 input1 # executable with notional input optionsParametric Sweep / HTC jobs
Consult the PyLauncher at TACC documentation for instructions on running parameter sweep and other High Throughput Computing workflows.
One Multi-Threaded Application
Launch a threaded application the same way. Be sure to specify the number of threads. Note that the default OpenMP thread count is 1.
export OMP_NUM_THREADS=128 # 128 total OpenMP threads (1 per core)
./myprogramOne MPI Application
To launch an MPI application, use the TACC-specific MPI launcher ibrun, which is a Lonestar6-aware replacement for generic MPI launchers like mpirun and mpiexec. In most cases the only arguments you need are the name of your executable followed by any arguments your executable needs. When you call ibrun without other arguments, your Slurm #SBATCH directives will determine the number of ranks (MPI tasks) and number of nodes on which your program runs.
#SBATCH -N 4
#SBATCH -n 512
# ibrun uses the $SBATCH directives to properly allocate nodes and tasks
ibrun ./myprogram To use ibrun interactively, say within an idev session, you can specify:
login1$ idev -N 2 -n 100
c309-005$ ibrun ./myprogramOne Hybrid (MPI+Threads) Application
When launching a single application you generally don't need to worry about affinity: both Intel MPI and MVAPICH2 will distribute and pin tasks and threads in a sensible way.
export OMP_NUM_THREADS=8 # 8 OpenMP threads per MPI rank
ibrun ./myprogram # use ibrun instead of mpirun or mpiexecAs a practical guideline, the product of $OMP_NUM_THREADS and the maximum number of MPI processes per node should not be greater than total number of cores available per node (128 cores in the development/normal/large queues).
More Than One Serial Application in the Same Job
TACC's pylauncher utility provides an easy way to launch more than one serial application in a single job. This is a great way to engage in a popular form of High Throughput Computing: running parameter sweeps (one serial application against many different input datasets) on several nodes simultaneously. The PyLauncher utility will execute your specified list of independent serial commands, distributing the tasks evenly, pinning them to specific cores, and scheduling them to keep cores busy. Consult PyLauncher at TACC for more information.
MPI Applications One at a Time
To run one MPI application after another (or any sequence of commands one at a time), simply list them in your job script in the order in which you'd like them to execute. When one application/command completes, the next one will begin.
./preprocess.sh
ibrun ./myprogram input1 # runs after preprocess.sh completes
ibrun ./myprogram input2 # runs after previous MPI app completesMore Than One MPI Application Running Concurrently
To run more than one MPI application simultaneously in the same job, you need to do several things:
- use ampersands to launch each instance in the background;
- include a wait command to pause the job script until the background tasks complete;
- use
ibrun's-nand-oswitches to specify task counts and hostlist offsets respectively; and - include a call to the
task_affinityscript in youribrunlaunch line.
If, for example, you use #SBATCH directives to request N=4 nodes and n=256 total MPI tasks, Slurm will generate a hostfile with 256 entries (64 entries for each of 4 nodes). The -n and -o switches, which must be used together, determine which hostfile entries ibrun uses to launch a given application; execute ibrun --help for more information. Don't forget the ampersands ("&") to launch the jobs in the background, and the wait command to pause the script until the background tasks complete:
# 128 tasks; offset by 0 entries in hostfile.
ibrun -n 128 -o 0 task_affinity ./myprogram input1 &
# 128 tasks; offset by 128 entries in hostfile.
ibrun -n 128 -o 128 task_affinity ./myprogram input2 &
# Required; else script will exit immediately.
waitThe task_affinity script manages task placement and memory pinning when you call ibrun with the -n, -o switches (it's not necessary under any other circumstances).
More than One OpenMP Application Running Concurrently
You can also run more than one OpenMP application simultaneously on a single node, but you will need to distribute and pin OpenMP threads appropriately. The most portable way to do this is with OpenMP Affinity.
An OpenMP executable sequentially assigns its N forked threads (thread number 0,...N-1) at a parallel region to the sequence of "places" listed in the $OMP_PLACES environment variable. Each place is specified within braces ({}). The sequence {0,1},{2,3},{4,5} has three places, and OpenMP thread numbers 0, 1, and 2 are assigned to the processor ids (proc-ids) 0,1 and 2,3 and 4,5, respectively. The hardware assigned to the proc-ids can be found in the /proc/cpuinfo file.
The sequence of proc-ids on socket 0 and socket 1 are sequentially numbered.
On socket 0:
0,1,2,...,62,63 and on socket 1:
64,65,...,126,127Note, hardware threads are not enabled on Lonestar6. So, there are no core ids greater than 127.
The proc-id mapping to the cores for Milan is:
|------- Socket 0 ------------|-------- Socket 1 -------------|
# 0 1 2,..., 61, 62, 63 | 0 1 2,..., 61, 62, 63 |
0 0 1 2,..., 61, 62, 63 | 64 65 66,..., 125, 126, 127 |Hence, to bind OpenMP threads to a sequence of 3 cores on each socket, the places would be:
socket 0: export OMP_PLACES="{0},{1},{2}"
socket 1: export OMP_PLACES="{64},{65},{66}"Under the NUMA covers, each AMD chip is actually composed of 8 "chiplets" which share a 32 MB L3 cache. To place each thread on its own chiplet for an 8 thread OpenMP program, you would use this command:
socket 0: export OMP_PLACES="{0},{8},{16},{24},{32},{40},{48},{56}"
socket 1: export OMP_PLACES="{64},{72},{80},{88},{96},{104},{112},{120}"Interval notation can be used to express a sequence of places. The syntax is: {proc-ids},N,S, where N is the number of places to create from the base place ({proc-ids}) with a stride of S. Hence the above sequences could have been written:
socket 0: export OMP_PLACES="{0},8,8"
socket 1: export OMP_PLACES="{64},8,8"In the example below two OpenMP programs are executed on a single node, each using 64 threads. The first program uses the cores on socket 0. It is put in the background, using the ampersand (&) character at the end of the line, so that the job script execution can continue to the second OpenMP program execution, which uses the cores on socket 1. It, too, is put in the background, and the job execution waits for both to finish with the wait command at the end.
export OMP_NUM_THREADS=64
env OMP_PLACES="{0},64,1" ./omp.exe & #execution on socket 0 cores
env OMP_PLACES="{64},64,1" ./omp.exe & #execution on socket 1 cores
waitRunning Jobs
Lonestar6's job scheduler is the Slurm Workload Manager. Slurm commands enable you to submit, manage, monitor, and control your jobs. Jobs submitted to the scheduler are queued, then run on the compute nodes. Each job consumes Service Units (SUs) which are then charged to your allocation. See the Job Management section below for further information.
Production Queues
Lonestar6's new queue, vm-small is designed for users who only need a subset of a node's entire 128 cores in the "normal" queue. Run your jobs in this queue if your job requires 16 cores or less and needs less than 29 GB of memory. If your job is memory bandwidth dependent, your performance may decrease since your job will be possibly sharing memory bandwidth with other jobs.
The jobs in this queue consume 1/7 the resources of a full node. Jobs are charged accordingly at .143 SUs per node hour.
Important
Queue limits are subject to change without notice.
Frontera admins may occasionally adjust queue settings in order to ensure fair scheduling for the entire user community.
TACC's qlimits utility will display the latest queue configurations.
Warning
Queue Restrictions Do not request specific nodes when submitting jobs without prior approval from staff. Allow Slurm to allocate nodes as appropriate.
Any job requesting specific compute nodes via batch scripts, idev invocations, or MPI hostfiles will be deleted from the queue.
Table 5. Production Queues
| Queue Name | Min/Max Nodes per Job (assoc'd cores)* |
Max Job Duration |
Max Nodes per User |
Max Jobs per User |
Max Submit | Charge Rate (per node-hour) |
|---|---|---|---|---|---|---|
development |
8 nodes (1024 cores) |
2 hours | 8 | 1 | 3 | 1 SU |
gpu-a100 |
8 nodes (1024 cores) |
48 hours | 12 | 8 | 32 | 3 SUs |
gpu-a100-dev |
2 nodes (256 cores) |
2 hours | 2 | 1 | 3 | 3 SUs |
gpu-a100-small** |
1 node | 48 hours | 3 | 3 | 12 | 1.5 SUs |
gpu-h100 |
1 node | 48 hours | 1 | 1 | 4 | 6 SUs |
large* |
65/256 nodes (65536 cores) |
48 hours | 256 | 1 | 4 | 1 SU |
normal |
1/64 nodes (8192 cores) |
48 hours | 64 | 20 | 100 | 1 SU |
vm-small** |
1 node (16 cores) |
48 hours | 4 | 4 | 16 | 0.143 SU |
* Access to the large queue is restricted. To request more nodes than are available in the normal queue, submit a consulting (help desk) ticket through the TACC User Portal. Include in your request reasonable evidence of your readiness to run under the conditions you're requesting. In most cases this should include your own strong or weak scaling results from Lonestar6.
** The gpu-a100-small and vm-small queues contain virtual nodes with fewer resources (cores) than the nodes in the other queues.
Job Accounting
Like all TACC systems, Lonestar6's accounting and allocation system is based on node-hours: one unadjusted Service Unit (SU) represents a single compute node used for one hour (a node-hour). For any given job, the total cost in SUs is the use of one compute node for one hour of wall clock time plus any charges or discounts for the use of specialized queues, e.g. Stampede3's pvc queue, Lonestar6's gpu-a100 queue, and Frontera's flex queue. The queue charge rates are determined by the supply and demand for that particular queue or type of node used and are subject to change.
The Slurm scheduler tracks and charges for usage to a granularity of a few seconds of wall clock time. The system charges only for the resources you actually use, not those you request. If your job finishes early and exits properly, Slurm will release the nodes back into the pool of available nodes. Your job will only be charged for as long as you are using the nodes.
TACC does not implement node-sharing on any compute resource. Each Lonestar6 compute node can be assigned to only one user at a time; hence a complete node is dedicated to a user's job and accrues wall-clock time for all the node's cores/GPUs whether or not all cores/GPUs are used.
TACC Charging Policy
Lonestar6 SUs billed = (# nodes) x max(job duration in wall clock hours,.25) x (charge rate per node-hour)
All running jobs are charged a minimum of 15 minutes (.25 hrs) of queue time regardless of actual runtime. This policy ensures equal access to the queues for all users as TACC's user base expands.
For example: a 4-node job in Lonestar6's gpu-a100 queue which runs for five minutes would be charged as follows:
4 nodes * 0.25 hrs * 3 SUs/node-hour = 3 SUs
We strongly encourage users launching large jobs to do thorough testing of your code at smaller node counts prior to maximizing your runs.
Submitting Batch Jobs with sbatch
Use Slurm's sbatch command to submit a batch job to one of the Lonestar6 queues:
login1$ sbatch myjobscriptWhere myjobscript is the name of a text file containing #SBATCH directives and shell commands that describe the particulars of the job you are submitting. The details of your job script's contents depend on the type of job you intend to run.
In your job script you (1) use #SBATCH directives to request computing resources (e.g. 10 nodes for 2 hrs); and then (2) use shell commands to specify what work you're going to do once your job begins. There are many possibilities: you might elect to launch a single application, or you might want to accomplish several steps in a workflow. You may even choose to launch more than one application at the same time. The details will vary, and there are many possibilities. But your own job script will probably include at least one launch line that is a variation of one of the examples described here.
Your job will run in the environment it inherits at submission time; this environment includes the modules you have loaded and the current working directory. In most cases you should run your applications(s) after loading the same modules that you used to build them. You can of course use your job submission script to modify this environment by defining new environment variables; changing the values of existing environment variables; loading or unloading modules; changing directory; or specifying relative or absolute paths to files. Do not use the Slurm --export option to manage your job's environment: doing so can interfere with the way the system propagates the inherited environment.
Table 5.5 below describes some of the most common sbatch command options. Slurm directives begin with #SBATCH; most have a short form (e.g. -N) and a long form (e.g. --nodes). You can pass options to sbatch using either the command line or job script; most users find that the job script is the easier approach. The first line of your job script must specify the interpreter that will parse non-Slurm commands; in most cases #!/bin/bash or #!/bin/csh is the right choice. Avoid #!/bin/sh (its startup behavior can lead to subtle problems on Lonestar6), and do not include comments or any other characters on this first line. All #SBATCH directives must precede all shell commands. Note also that certain #SBATCH options or combinations of options are mandatory, while others are not available on Lonestar6.
By default, Slurm writes all console output to a file named "slurm-%j.out", where %j is the numerical job ID. To specify a different filename use the -o option. To save stdout (standard out) and stderr (standard error) to separate files, specify both -o and -e options.
Tip
The maximum runtime for any individual job is 48 hours. However, if you have good checkpointing implemented, you can easily chain jobs such that the outputs of one job are the inputs of the next, effectively running indefinitely for as long as needed. See Slurm's -d option.
Table 5.5 Common sbatch Options
| Option | Argument | Comments |
|---|---|---|
-A |
projectid | Charge job to the specified project/allocation number. This option is only necessary for logins associated with multiple projects. |
-aor --array |
=tasklist | Lonestar6 supports Slurm job arrays. See the Slurm documentation on job arrays for more information. |
-d= |
afterok:jobid | Specifies a dependency: this run will start only after the specified job (jobid) successfully finishes |
-export= |
N/A | Avoid this option on Lonestar6. Using it is rarely necessary and can interfere with the way the system propagates your environment. |
--gres |
N/A | Lonestar6 does not support this option. Slurm will reject any job script containing this directive. |
--gpus-per-task |
N/A | Lonestar6 does not support this option. Slurm will reject any job script containing this directive. |
-p |
queue_name | Submits to queue (partition) designated by queue_name |
-J |
job_name | Job Name |
-N |
total_nodes | Required. Define the resources you need by specifying either: (1) -N and -n; or(2) -N and -ntasks-per-node. |
-n |
total_tasks | This is total MPI tasks in this job. See -N above for a good way to use this option. When using this option in a non-MPI job, it is usually best to set it to the same value as -N. |
-ntasks-per-nodeor -tasks-per-node |
tasks_per_node | This is MPI tasks per node. See -N above for a good way to use this option. When using this option in a non-MPI job, it is usually best to set -ntasks-per-node to 1. |
-t |
hh:mm:ss | Required. Wall clock time for job. |
-mail-type= |
begin, end, fail, or all |
Specify when user notifications are to be sent (one option per line). |
-mail-user= |
email_address | Specify the email address to use for notifications. Use with the -mail-type= flag above. |
-o |
output_file | Direct job standard output to output_file (without -e option error goes to this file) |
-e |
error_file | Direct job error output to error_file |
-mem |
N/A | Not available. If you attempt to use this option, the scheduler will not accept your job. |
Sample Job Scripts
Copy and customize the following scripts to specify and refine your job's requirements.
- specify the maximum run time with the
-toption. - specify number of nodes needed with the
-Noption - specify total number of MPI tasks with the
-noption - specify the project to be charged with the
-Aoption.
In general, the fewer resources (nodes) you specify in your batch script, the less time your job will wait in the queue. See 5. Job Submissions Tips in the Good Conduct document.
Consult Table 9 in the Stampede3 User Guide for a listing of common Slurm #SBATCH options.
Click on a tab header below to display it's job script, then copy and customize to suit your own application.
Serial Jobs
Serial codes should request 1 node (#SBATCH -N 1) with 1 task (#SBATCH -n 1).
Important
Run all serial jobs in the normal queue.
Consult the PyLauncher at TACC documentation to run multiple serial executables at one time.
#!/bin/bash
#----------------------------------------------------
# Sample Slurm job script
# for TACC Lonestar6 AMD Milan nodes
#
# *** Serial Job in Normal Queue***
#
# Last revised: October 22, 2021
#
# Notes:
#
# -- Copy/edit this script as desired. Launch by executing
# "sbatch milan.serial.slurm" on a Lonestar6 login node.
#
# -- Serial codes run on a single node (upper case N = 1).
# A serial code ignores the value of lower case n,
# but slurm needs a plausible value to schedule the job.
#
# -- Use TACC's pylauncher utility to run multiple serial
# executables at the same time, execute "module load pylauncher"
# followed by "module help pylauncher".
#----------------------------------------------------
#SBATCH -J myjob # Job name
#SBATCH -o myjob.o%j # Name of stdout output file
#SBATCH -e myjob.e%j # Name of stderr error file
#SBATCH -p normal # Queue (partition) name
#SBATCH -N 1 # Total # of nodes (must be 1 for serial)
#SBATCH -n 1 # Total # of mpi tasks (should be 1 for serial)
#SBATCH -t 01:30:00 # Run time (hh:mm:ss)
#SBATCH --mail-type=all # Send email at begin and end of job
#SBATCH -A myproject # Project/Allocation name (req'd if you have more than 1)
#SBATCH --mail-user=username@tacc.utexas.edu
# Any other commands must follow all #SBATCH directives...
module list
pwd
date
# Launch serial code...
./myprogram # Do not use ibrun or any other MPI launcherMPI Jobs
This job script requests 4 nodes (#SBATCH -N 4) and 32 tasks (#SBATCH -n 32), for 8 MPI ranks per node.
#!/bin/bash
#----------------------------------------------------
# Sample Slurm job script
# for TACC Lonestar6 AMD Milan nodes
#
# *** MPI Job in Normal Queue ***
#
# Last revised: October 22, 2021
#
# Notes:
#
# -- Launch this script by executing
# "sbatch milan.mpi.slurm" on a Lonestar6 login node.
#
# -- Use ibrun to launch MPI codes on TACC systems.
# Do NOT use mpirun or mpiexec.
#
# -- Max recommended MPI ranks per Milan node: 128
# (start small, increase gradually).
#
# -- If you're running out of memory, try running
# fewer tasks per node to give each task more memory.
#
#----------------------------------------------------
#SBATCH -J myjob # Job name
#SBATCH -o myjob.o%j # Name of stdout output file
#SBATCH -e myjob.e%j # Name of stderr error file
#SBATCH -p normal # Queue (partition) name
#SBATCH -N 4 # Total # of nodes
#SBATCH -n 32 # Total # of mpi tasks
#SBATCH -t 01:30:00 # Run time (hh:mm:ss)
#SBATCH --mail-type=all # Send email at begin and end of job
#SBATCH -A myproject # Project/Allocation name (req'd if you have more than 1)
#SBATCH --mail-user=username@tacc.utexas.edu
# Any other commands must follow all #SBATCH directives...
module list
pwd
date
# Launch MPI code...
ibrun ./myprogram # Use ibrun instead of mpirun or mpiexecOpenMP Jobs
Important
Run all OpenMP jobs in the normal queue.
#!/bin/bash
#----------------------------------------------------
# Sample Slurm job script
# for TACC Lonestar6 AMD Milan nodes
#
# *** OpenMP Job in Normal Queue ***
#
# Last revised: October 22, 2021
#
# Notes:
#
# -- Launch this script by executing
# -- Copy/edit this script as desired. Launch by executing
# "sbatch milan.openmp.slurm" on a Lonestar6 login node.
#
# -- OpenMP codes run on a single node (upper case N = 1).
# OpenMP ignores the value of lower case n,
# but slurm needs a plausible value to schedule the job.
#
# -- Default value of OMP_NUM_THREADS is 1; be sure to change it!
#
# -- Increase thread count gradually while looking for optimal setting.
# If there is sufficient memory available, the optimal setting
# is often 56 (1 thread per core) but may be higher.
#----------------------------------------------------
#SBATCH -J myjob # Job name
#SBATCH -o myjob.o%j # Name of stdout output file
#SBATCH -e myjob.e%j # Name of stderr error file
#SBATCH -p normal # Queue (partition) name
#SBATCH -N 1 # Total # of nodes (must be 1 for OpenMP)
#SBATCH -n 1 # Total # of mpi tasks (should be 1 for OpenMP)
#SBATCH -t 01:30:00 # Run time (hh:mm:ss)
#SBATCH --mail-type=all # Send email at begin and end of job
#SBATCH --mail-user=username@tacc.utexas.edu
#SBATCH -A myproject # Project/Allocation name (req'd if you have more than 1)
# Any other commands must follow all #SBATCH directives...
module list
pwd
date
# Set thread count (default value is 1)...
export OMP_NUM_THREADS=56 # this is 1 thread/core; may want to start lower
# Launch OpenMP code...
./myprogram # Do not use ibrun or any other MPI launcherHybrid (MPI + OpenMP) Jobs
This script requests 10 nodes (#SBATCH -N 10) and 40 tasks (#SBATCH -n 40).
#!/bin/bash
#----------------------------------------------------
# Example Slurm job script
# for TACC Lonestar6 AMD Milan nodes
#
# *** Hybrid Job in Normal Queue ***
#
# This sample script specifies:
# 10 nodes (capital N)
# 40 total MPI tasks (lower case n); this is 4 tasks/node
# 14 OpenMP threads per MPI task (56 threads per node)
#
# Last revised: October 22, 2021
#
# Notes:
#
# -- Launch this script by executing
# "sbatch milan.hybrid.slurm" on Lonestar6 login node.
#
# -- Use ibrun to launch MPI codes on TACC systems.
# Do NOT use mpirun or mpiexec.
#
# -- In most cases it's best to keep
# ( MPI ranks per node ) x ( threads per rank )
# to a number no more than 56 (total cores).
#
# -- If you're running out of memory, try running
# fewer tasks and/or threads per node to give each
# process access to more memory.
#
# -- IMPI does sensible process pinning by default.
#
#----------------------------------------------------
#SBATCH -J myjob # Job name
#SBATCH -o myjob.o%j # Name of stdout output file
#SBATCH -e myjob.e%j # Name of stderr error file
#SBATCH -p normal # Queue (partition) name
#SBATCH -N 10 # Total # of nodes
#SBATCH -n 40 # Total # of mpi tasks
#SBATCH -t 01:30:00 # Run time (hh:mm:ss)
#SBATCH --mail-type=all # Send email at begin and end of job
#SBATCH -A myproject # Project/Allocation name (req'd if you have more than 1)
#SBATCH --mail-user=username@tacc.utexas.edu
# Any other commands must follow all #SBATCH directives...
module list
pwd
date
# Set thread count (default value is 1)...
export OMP_NUM_THREADS=14
# Launch MPI code...
ibrun ./myprogram # Use ibrun instead of mpirun or mpiexecJob Management
In this section, we present several Slurm commands and other utilities that are available to help you plan and track your job submissions as well as check the status of the Slurm queues.
Important
When interpreting queue and job status, remember that Lonestar6 does not operate on a first-come-first-served basis. Instead, the sophisticated, tunable algorithms built into Slurm attempt to keep the system busy, while scheduling jobs in a way that is as fair as possible to everyone. At times this means leaving nodes idle ("draining the queue") to make room for a large job that would otherwise never run. It also means considering each user's "fair share", scheduling jobs so that those who haven't run jobs recently may have a slightly higher priority than those who have.
Monitoring Queue Status
Monitor queue status via Slurm's sinfo command and/or TACC's qlimits utility.
TACC's qlimits command
To display resource limits for the Lonestar queues, execute: qlimits. The result is real-time data; the corresponding information in this document's table of Lonestar6 queues may lag behind the actual configuration that the qlimits utility displays.
Slurm's sinfo command
Slurm's sinfo command allows you to monitor the status of the queues. If you execute sinfo without arguments, you'll see a list of every node in the system together with its status. To skip the node list and produce a tight, alphabetized summary of the available queues and their status, execute:
login1$ sinfo -S+P -o "%18P %8a %20F" # compact summary of queue statusPARTITION AVAIL NODES(A/I/O/T)
corralextra up 512/0/1/513
corralextra-dev up 18/0/0/18
corralextra-gpu up 72/1/0/73
development* up 18/0/0/18
gpu-a100 up 72/1/0/73
gpu-a100-dev up 4/0/0/4
gpu-a100-small up 14/10/0/24
gpu-h100 up 2/2/0/4
large up 512/0/1/513
normal up 512/0/1/513
vm-small up 28/0/0/28The AVAIL column displays the overall status of each queue (up or down), while the column labeled NODES(A/I/O/T) shows the number of nodes in each of several states ("Allocated", "Idle", "Offline", and "Total"). Execute man sinfo for more information. Use caution when reading the generic documentation, however: some available fields are not meaningful or are misleading on Lonestar6 (e.g. TIMELIMIT, displayed using the %l option).
Monitoring Job Status
Monitor your job's status in the queue via Slurm's squeue command and/or TACC's showq utility.
Slurm's squeue command
Slurm's squeue command displays the state of all queued and running jobs.
login1$ squeue # show all jobs in all queues
login1$ squeue -u bjones # show all jobs owned by bjones
login1$ man squeue # more infoThe squeue command's default output format lists all nodes assigned to displayed jobs; this can make the output difficult to read. See the example below for a handy variation that suppresses the nodelist:
Tip
The squeue command's default output format lists all nodes assigned to displayed jobs; this can make the output difficult to read. See the example below for a handy variation that suppresses the nodelist:
login1$ squeue -o "%.10i %.12P %.12j %.9u %.2t %.9M %.6D" # suppress nodelistJob and Queue Status Meanings
The squeue command's output displays two columns of interest, the column labeled ST displays each job's status, and the last column, labeled NODELIST/REASON, includes a nodelist for running/completing jobs, or a reason for pending jobs. See Table 6 and Table 7 below for detailed explanations.
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
3146313 development st_archi oxygen PD 0:00 1 (Dependency)
3148608 development HETDEX ecooper R 29:05 4 c307-[005-006],c308-[005-006]
3148393 development rhoCentr subhajit R 1:39:30 2 c305-[005-006]
3148334 gpu-a100 0.25_Li3 clp19 PD 0:00 1 (QOSMaxJobsPerUserLimit)
3148337 gpu-a100 0.25_Li3 clp19 PD 0:00 1 (QOSMaxJobsPerUserLimit)
3141553 gpu-a100 WOY yanghj20 R 21:47:55 1 c316-001
3148264 gpu-a100 ML_GC_ja mukherje R 2:39:00 1 c316-010
3146770 gpu-a100-sm inpaint_ yantingx R 18:28:29 1 v330-023
3145353 gpu-a100-sm RF_DQN_j mukherje R 1-10:41:37 1 v330-001
3140472 gpu-h100 train zxruan R 1-06:00:32 1 c318-004
3147044 gpu-h100 idv66384 singhsud R 3:48:51 1 c318-001
3142355 normal nvt-pack zbajalan PD 0:00 1 (QOSMaxJobsPerUserLimit)
3142353 normal nvt-pack zbajalan PD 0:00 1 (QOSMaxJobsPerUserLimit)
3148240 normal neid_202 jplneid R 2:40:46 1 c314-053
3142640 normal POLARIS kmori R 1-19:59:11 1 c312-214
3146504 vm-small 8sigGNP pc104 PD 0:00 1 (QOSMaxJobsPerUserLimit)
3148638 vm-small A2 nemian PD 0:00 1 (Resources)
3148649 vm-small MULTIPAR rma0798 PD 0:00 1 (Priority)
3148446 vm-small rhoCentr subhajit R 46:17 1 v320-016squeue outputTable 6. Job Status Meanings
| Status Code | Status | Description |
|---|---|---|
CA |
CANCELLED | Job was explicitly cancelled by the user or system administrator |
CD |
COMPLETED | Job has terminated all processes on all nodes with an exit code of zero. |
CG |
COMPLETING | Job is in the process of completing. Some processes on some nodes may still be active. |
F |
FAILED | Job terminated with non-zero exit code or other failure condition. |
NF |
NODE_FAIL | Job terminated due to failure of one or more allocated nodes. |
PD |
PENDING | Job is awaiting resource allocation. |
PR |
PREEMPTED | Job terminated due to preemption. |
R |
RUNNING | Job has an allocation and is currently running. |
TO |
TIMEOUT | Job terminated upon reaching its time limit. |
Table 7. Pending Jobs Reason
The last column, labeled NODELIST/REASON, includes a nodelist for running/completing jobs, or a reason for pending jobs.
NODELIST/REASON |
Description |
|---|---|
Resources |
The necessary combination of nodes/GPUs for your job are not available |
Priority |
There are other jobs in the queue with a higher priority |
Dependency |
The job will not start until the dependency specified by you is satisfied. |
ReqNodeNotAvailable |
If you submit a job before a scheduled system maintenance period, and the job cannot complete before the maintenance begins, your job will run when the maintenance/reservation concludes. The job will remain in the PD state until Lonestar6 returns to production. |
QOSMaxJobsPerUserLimit |
The number of your jobs queued exceeds that queue's limits. These jobs will run once your previous jobs have ended. |
Tip
The --start option to the squeue command displays job start times, including very rough estimates for the expected start times of some pending jobs that are relatively high in the queue:
login1$ squeue --start -j 167635 # display estimated start time for job 167635TACC's showq utility
TACC's showq utility mimics a tool that originated in the PBS project, and serves as a popular alternative to the Slurm squeue command:
login1$ showq # show all jobs; default format
login1$ showq -u # show your own jobs
login1$ showq -U bjones # show jobs associated with user bjones
login1$ showq -h # more infoThe output groups jobs in four categories: ACTIVE, WAITING, BLOCKED, and COMPLETING/ERRORED. A BLOCKED job is one that cannot yet run due to temporary circumstances (e.g. a pending maintenance or other large reservation.).
If your waiting job cannot complete before a maintenance/reservation begins, showq will display its state as **WaitNod** ("Waiting for Nodes"). The job will remain in this state until Lonestar6 returns to production.
Since TACC charges by the node rather than core, showq's default format now reports total nodes associated with a job rather than cores, tasks, or hardware threads. Run showq with the -l option to display the number of cores and the job's queue.
Dependent Jobs using sbatch
You can use sbatch to help manage workflows that involve multiple steps: the --dependency option allows you to launch jobs that depend on the completion (or successful completion) of another job. For example you could use this technique to split into three jobs a workflow that requires you to (1) compile on a single node; then (2) compute on 40 nodes; then finally (3) post-process your results using 4 nodes.
login1$ sbatch --dependency=afterok:173210 myjobscriptWarning
It is not possible to add resources to a job (e.g. allow more time, increase number of nodes) once you've submitted the job to the queue.
For more information see the Slurm online documentation. Note that you can use $SLURM_JOBID from one job to find the jobid you'll need to construct the sbatch launch line for a subsequent one. But also remember that you can't use sbatch to submit a job from a compute node.
Inspecting Running and Completed Jobs
-
To view some accounting data associated with your own jobs, use
sacct:login1$ sacct --starttime 2026-05-01 # show jobs that started on or after this date -
To cancel a pending or running job, first determine its jobid, then use
scancel:login1$ squeue -u bjones # one way to determine jobid JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON) 170361 v100 spec12 bjones PD 0:00 32 (Resources) login1$ scancel 170361 # cancel job -
For detailed information about the configuration of a specific job, use
scontrol:login1$ scontrol show job=170361
Machine Learning on LS6
You may utilize one or more nodes for machine-learning model training tasks on Lonestar6. Lonestar6 supports both configurations, enabling researchers to scale their training efficiently based on project requirements.
- Single-node training utilizes one or multiple GPUs on a single node and is ideal for smaller models and datasets, offering faster job throughput and easier debugging.
- Multi-node training, on the other hand, distributes the workload across multiple nodes, each with its own set of GPUs. This approach is necessary for large-scale models and datasets that exceed the memory or compute capacity of a single node, and is essential for scalability.
Environment Setup
Before diving into machine learning frameworks, it's essential to set up your environment correctly. Lonestar6 supports Python virtual environments, which help manage dependencies and isolate projects. This guide walks you through setting up environments for both PyTorch and Accelerate.
We recommend using a Python virtual environment to manage machine learning packages. Once set up, users can run jobs within this environment using either idev sessions or SLURM batch scripts.
PyTorch
Follow these step to set up PyTorch on Lonestar6 within a Python virtual environment.
Important
Install your python virtual environments on Lonestar6's $SCRATCH, file system. Do not use TACC's $WORK file system.
-
Request a single compute node in Lonestar6's
gpu-a100-devqueue using TACC'sidev utility:login1$ idev -p gpu-a100-dev -N 1 -n 1 -t 1:00:00 -
Create a Python virtual environment:
c123-456$ module load python3/3.9.7 c123-456$ python3 -m venv /path/to/virtual-env # (e.g., $SCRATCH/python-envs/test) -
Activate the Python virtual environment:
c123-456$ source /path/to/virtual-env/bin/activate -
Now install PyTorch:
c123-456$ pip3 install torch==2.8.0 torchvision torchaudio --index-url https://download.pytorch.org/whl/test/cu128
Testing PyTorch Installation
To test your installation of PyTorch we point you to a few benchmark calculations that are part of PyTorch's tutorials on multi-GPU and multi-node training. See PyTorch's documentation:
Distributed Data Parallel in PyTorch. These tutorials include several scripts set up to run single-node training and multi-node training.
Single-Node
-
Download the benchmark:
c123-456$ cd $SCRATCH c123-456$ git clone https://github.com/pytorch/examples.git -
Run the benchmark on one node (3 GPUs):
c123-456$ torchrun --nproc_per_node=3 examples/distributed/ddp-tutorial-series/multigpu_torchrun.py 50 10
Multi-Node
-
Request two nodes in the gpu-a100-dev queue using TACC's idev utility:
login1$ idev -N 2 -n 2 -p gpu-a100-dev -t 01:00:00 -
Move to the benchmark directory:
c123-456$ cd $SCRATCH -
Create a script called "run.sh". This script needs two parameters, the hostname of the master node and the number of nodes. Add execution permission for the file "run.sh".
#!/bin/bash HOST=$1 NODES=$2 LOCAL_RANK=${PMI_RANK} torchrun --nproc_per_node=3 --nnodes=$NODES --node_rank=${LOCAL_RANK} --master_addr=$HOST \ examples/distributed/ddp-tutorial-series/multinode.py 50 10 -
Run multi-gpu training:
c123-456$ ibrun -np 2 ./run.sh c123-456 2
Transformers with Accelerate
Transformers is a Python library developed by Hugging Face that provides pre-trained models for machine learning tasks. It includes implementations of popular architectures like BERT, GPT, and T5, making it easy to fine-tune and deploy state-of-the-art models.
Accelerate is a Hugging Face library designed to streamline distributed training. It abstracts away the complexity of launching multi-GPU and multi-node jobs, allowing users to scale their training with minimal code changes. Follow these steps to set up and run training jobs.
Set up Environment for Transformers
- Download the code and scripts for the job and change directory
c123-456$ cd $SCRATCH c123-456$ git clone https://github.com/skye-glitch/Machine-Learning-on-LS6.git c123-456$ cd Machine-Learning-on-LS6 -
Request a single compute node in Lonestar6's
gpu-a100-devqueue using TACC'sidevutility:login1$ idev -p gpu-a100-dev -N 1 -n 1 -t 1:00:00 -
Create a Python virtual environment:
c123-456$ module load python3/3.9.7 c123-456$ python3 -m venv .venv -
Request a single compute node in Lonestar6's gpu-a100-dev queue using TACC's idev utility:
login1$ idev -p gpu-a100-dev -N 1 -n 1 -t 1:00:00 -
Activate the Python virtual environment:
c123-456$ source .venv -
Now install dependencies:
c123-456$ pip3 install -r requirements.txt
Testing Transformers Installation
Single-Node
Run the training on one node (3 GPUs):
c123-456$ accelerate launch --num_processes 3 ./finetune.pyMulti-Node
-
Request two nodes in the gpu-a100-dev queue using TACC's idev utility:
login2.ls6$ idev -N 2 -n 2 -p gpu-a100-dev -t 01:00:00 -
Move to the code directory:
c123-456$ cd $SCRATCH/Machine-Learning-on-LS6 -
Run the multi-gpu training:
c123-456$ mpirun -np 2 --map-by ppr:1:node multinode.sh
Visualization and VNC Sessions
Lonestar6 uses AMD's Milan processors for all visualization and rendering operations. We use the Intel OpenSWR library to render raster graphics with OpenGL, and the Intel OSPRay framework for ray traced images inside visualization software. OpenSWR can be loaded by executing module load swr.
Lonestar6 currently has no separate visualization queue. All visualization apps are available on all nodes. VNC and DCV sessions are available on any queue, either through the command line or via the TACC Analysis Portal. We recommend submitting to Lonestar6's development queue for interactive sessions. If you are interested in an application that is not yet available, please submit a help desk ticket.
Remote Desktop Access
Remote desktop access to Lonestar6 is formed through a DCV or VNC connection to one or more compute nodes. Users must first connect to a Lonestar6 login node (see Accessing the System) and submit a special interactive batch job that:
- allocates a set of Lonestar6 compute nodes
- starts a
dcvserverorvncserverremote desktop process on the first allocated node - sets up a tunnel through the login node to the dcvserver or vncserver access port
Once the remote desktop process is running on the compute node and a tunnel through the login node is created, an output message identifies the access port for connecting a remote desktop viewer. A remote desktop viewer application is run on the user's remote system and presents the desktop to the user.
Important
If this is your first time connecting to Lonestar6 using VNC, you must run vncpasswd to create a password for your VNC servers. This should NOT be your login password! This mechanism only deters unauthorized connections; it is not fully secure, as only the first eight characters of the password are saved.
All VNC connections are tunneled through SSH for extra security, as described below.
Follow the steps below to start an interactive session.
-
Start a Remote Desktop
TACC has provided a DCV job script (
/share/doc/slurm/job.dcv), a VNC job script (/share/doc/slurm/job.vnc) and a combined job script that prefers DCV and fails over to VNC if a DCV license is not available (/share/doc/slurm/job.dcv2vnc). Each script requests one node in the development queue for two hours, creating a remote desktop session, either DCV or VNC.login1$ sbatch /share/doc/slurm/job.vnc login1$ sbatch /share/doc/slurm/job.dcv login1$ sbatch /share/doc/slurm/job.dcv2vncYou may modify or overwrite script defaults with sbatch command-line options. Note that the command options must be placed between
sbatchand the script:-t hours:minutes:secondsmodify the job runtime-A projectnumberspecify the project/allocation to be charged-N nodesspecify number of nodes needed-p partitionspecify an alternate queue
Consult Table 6 in the Stampede3 User Guide for a listing of common Slurm
#SBATCHoptions.All arguments after the job script name are sent to the vncserver command. For example, to set the desktop resolution to 1440x900, use:
login1$ sbatch /share/doc/slurm/job.vnc -geometry 1440x900The
vnc.jobscript starts avncserverprocess and writes to the output file,vncserver.outin the job submission directory, with the connect port for the vncviewer.Note that the DCV viewer adjusts desktop resolution to your browser or DCV client, so desktop resolution does not need to be specified.
Watch for the "To connect" message at the end of the output file, or watch the output stream in a separate window with the commands:
login1$ touch vncserver.out ; tail -f vncserver.out login1$ touch dcvserver.out ; tail -f dcvserver.outThe lightweight window manager,
xfce, is the default DCV and VNC desktop and is recommended for remote performance. Gnome is available; to use gnome, open the~/.vnc/xstartupfile (created after your first VNC session) and replacestartxfce4withgnome-session. Note that gnome may lag over slow internet connections. -
Create an SSH Tunnel to Lonestar6
DCV connections are encrypted via TLS and are secure. For VNC connections, TACC requires users to create an SSH tunnel from the local system to the Lonestar6 login node to assure that the connection is secure. The tunnels created for the VNC job operate only on the
localhostinterface, so you must uselocalhostin the port forward argument, not the Lonestar6 hostname. On a Unix or Linux system, execute the following command once the port has been opened on the Lonestar6 login node:localhost$ ssh -f -N -L xxxx:localhost:yyyy username@ls6.tacc.utexas.eduwhere:
yyyyis the port number given by the vncserver batch jobxxxxis a port on the remote system. Generally, the port number specified on the Lonestar6 login node,yyyy, is a good choice to use on your local system as well-finstructs SSH to only forward ports, not to execute a remote command-Nputs the ssh command into the background after connecting-Lforwards the port
On Windows systems find the menu in the Windows SSH client where tunnels can be specified, and enter the local and remote ports as required, then ssh to Lonestar6.
-
Connecting the vncviewer
Once the SSH tunnel has been established, use a VNC client to connect to the local port you created, which will then be tunneled to your VNC server on Lonestar6. Connect to localhost:xxxx, where xxxx is the local port you used for your tunnel. In the examples above, we would connect the VNC client to
localhost::xxxx. (Some VNC clients acceptlocalhost:xxxx).We recommend the TigerVNC VNC Client, a platform independent client/server application.
Once the desktop has been established, two initial xterm windows are presented (which may be overlapping). One, which is white-on-black, manages the lifetime of the VNC server process. Killing this window (typically by typing
exitorctrl-Dat the prompt) will cause the vncserver to terminate and the original batch job to end. Because of this, we recommend that this window not be used for other purposes; it is just too easy to accidentally kill it and terminate the session.The other xterm window is black-on-white, and can be used to start both serial programs running on the node hosting the vncserver process, or parallel jobs running across the set of cores associated with the original batch job. Additional xterm windows can be created using the window-manager left-button menu.
Applications on the Remote Desktop
From an interactive desktop, applications can be run from icons or from xterm command prompts. Two special cases arise: running parallel applications, and running applications that use OpenGL.
Parallel Applications from the Desktop
Parallel applications are run on the desktop using the same ibrun wrapper described above (see Running). The command:
c301-001$ ibrun ibrunoptions application applicationoptionswill run application on the associated nodes, as modified by the ibrun options.
OpenGL/X Applications On The Desktop
Lonestar6 uses the OpenSWR OpenGL library to perform efficient rendering. At present, the compute nodes on Lonestar6 do not support native X instances. All windowing environments should use a DCV desktop launched via the job script in /share/doc/slurm/job.dcv, a VNC desktop launched via the job script in /share/doc/slurm/job.vnc or using the TACC Analysis Portal.
swr: To access the accelerated OpenSWR OpenGL library, it is necessary to use the swr module to point to the swr OpenGL implementation and configure the number of threads to allocate to rendering.
c301-001$ module load swr
c301-001$ swr options application application-argsParallel VisIt on Lonestar6
VisIt was compiled under the GNU compiler and the MVAPICH2 and MPI stacks.
After connecting to a VNC server on Lonestar6, as described above, load the VisIt module at the beginning of your interactive session before launching the VisIt application:
c301-001$ module load visit
c301-001$ visitswr module. The software rendering libraries and environment provided by the swr module are built in to the VisIt module on LS6.
VisIt first loads a dataset and presents a dialog allowing for selecting either a serial or parallel engine. Select the parallel engine. Note that this dialog will also present options for the number of processes to start and the number of nodes to use; these options are actually ignored in favor of the options specified when the VNC server job was started.
Preparing Data for Parallel Visit
VisIt reads nearly 150 data formats. Except in some limited circumstances (particle or rectilinear meshes in ADIOS, basic netCDF, Pixie, OpenPMD and a few other formats), VisIt piggy-backs its parallel processing off of whatever static parallel decomposition is used by the data producer. This means that VisIt expects the data to be explicitly partitioned into independent subsets (typically distributed over multiple files) at the time of input. Additionally, VisIt supports a metadata file (with a .visit extension) that lists multiple data files of any supported format that hold subsets of a larger logical dataset. VisIt also supports a "brick of values (bov) format which supports a simple specification for the static decomposition to use to load data defined on rectilinear meshes. For more information on importing data into VisIt, see Getting Data Into VisIt.
Parallel ParaView on Lonestar6
After connecting to a VNC server on Lonestar6, as described above, do the following:
-
Set up your environment with the necessary modules:
c301-001$ module load intel/19 impi qt5/5.14.2 oneapi_rk/2021.4.0 swr/21.2.5 paraview/5.10.0 -
Launch ParaView:
c301-001$ swr -p 1 paraview [paraview client options] -
Click the "Connect" button, or select File -> Connect
-
Select the "auto" configuration, then press "Connect". In the Paraview Output Messages window, you'll see what appears to be an 'lmod' error, but can be ignored. Then you'll see the parallel servers being spawned and the connection established.
Help Desk
Important
Submit a help desk ticket at any time via the TACC User Portal. Be sure to include "Lonestar6" in the Resource field.
TACC Consulting operates from 8am to 5pm CST, Monday through Friday, except for holidays. Help the consulting staff help you by following these best practices when submitting tickets.
-
Do your homework before submitting a help desk ticket. What does the user guide and other documentation say? Search the internet for key phrases in your error logs; that's probably what the consultants answering your ticket are going to do. What have you changed since the last time your job succeeded?
-
Describe your issue as precisely and completely as you can: what you did, what happened, verbatim error messages, other meaningful output.
Tip
When appropriate, include as much meta-information about your job and workflow as possible including:
- directory containing your build and/or job script
- all modules loaded
- relevant job IDs
- any recent changes in your workflow that could affect or explain the behavior you're observing.
-
Subscribe to Lonestar6 User News. This is the best way to keep abreast of maintenance schedules, system outages, and other general interest items.
-
Have realistic expectations. Consultants can address system issues and answer questions about Lonestar6. But they can't teach parallel programming in a ticket, and may know nothing about the package you downloaded. They may offer general advice that will help you build, debug, optimize, or modify your code, but you shouldn't expect them to do these things for you.
-
Be patient. It may take a business day for a consultant to get back to you, especially if your issue is complex. It might take an exchange or two before you and the consultant are on the same page. If the admins disable your account, it's not punitive. When the file system is in danger of crashing, or a login node hangs, they don't have time to notify you before taking action.